New research found that a protein called FTL1 is the primary driver of brain aging and discovered that removing it actually reverses memory loss

Memory loss has been treated as one of the most inevitable things that happens to a human being. You get older, your brain slows down, you start losing words and names and threads of conversations, and medicine offers you ways to delay the process but nothing that actually turns it around. That framing just took a significant hit.
Researchers at UC San Francisco combed through every protein and gene that changes in the hippocampus as mice age. Out of everything that shifted between young and old animals, one protein stood out above everything else as consistently different. They called it FTL1. And when they removed it from the brains of old mice, the animals did not just stop declining. They recovered.
“It is truly a reversal of impairments,” said Saul Villeda, associate director of the UCSF Bakar Aging Research Institute and senior author of the study. “It’s much more than merely delaying or preventing symptoms.”
What FTL1 Is and Why Nobody Was Watching It
FTL1 stands for ferritin light chain 1. Ferritin is an iron storage protein, the kind of molecule that gets measured in routine bloodwork when doctors want to check your iron levels. It stores iron in cells throughout the body, including in the brain, and under normal circumstances this is a useful function. The brain needs iron for dozens of metabolic processes and ferritin keeps it accessible without letting it float freely where it could cause oxidative damage.
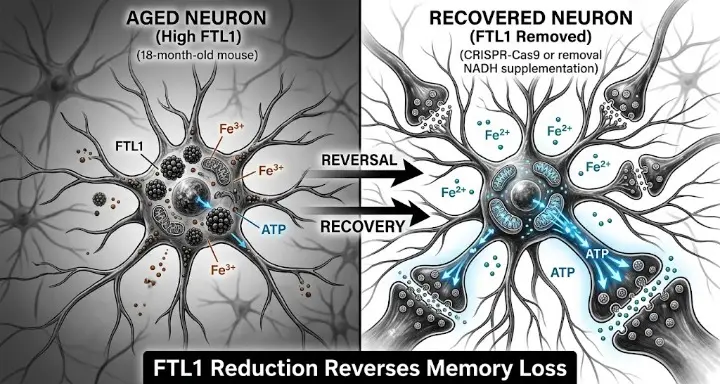
The problem begins when too much ferritin accumulates in neurons. The researchers detected an increase in neuronal FTL1 in the hippocampus of aged mice, the levels of which correlated with cognitive decline. This was not a modest association. When the UCSF team used viral vectors to artificially increase FTL1 in young three-month-old mice, those animals began showing the brain characteristics of old age almost immediately. Their neurons grew simple, single-armed extensions instead of the complex branching structures that healthy young brains produce. Their synaptic density dropped. Their memory performance on behavioral tests declined. Young mice with too much FTL1 looked, at the cellular level, like old mice.
Then the researchers did the opposite. They used both CRISPR-Cas9 and short hairpin RNA to reduce FTL1 in the hippocampi of mice aged 18 to 24 months. Their brain cells formed more connections, and they performed significantly better on memory tests. “The mice regained their youth,” explained Villeda.
The Iron Trap in Your Neurons
The mechanism the UCSF team uncovered runs through iron and energy. When FTL1 accumulates in aging neurons, it changes how iron exists inside the cell. Instead of remaining in a usable form, iron shifts toward an oxidized state, Fe3+, that the mitochondria cannot efficiently use. And mitochondria need iron specifically for building the iron-sulfur clusters that power their electron transport chain, the process through which they produce ATP.
“Our interpretation is that excess ferritin traps iron that mitochondria need for iron-sulfur clusters,” said Laura Remesal, the postdoctoral researcher who led the experimental work. “That lowers ATP and energy availability.”
Less ATP means neurons cannot maintain their synaptic connections. Synapses require constant energy to stay functional, to release neurotransmitters, to respond to incoming signals, to strengthen through the long-term potentiation process that is the cellular basis of learning and memory. When the energy supply drops, synapses weaken and eventually disappear. The neuron does not die. It just goes quiet, losing the connections that made it useful.
The chain the UCSF team documented goes: elevated FTL1, oxidized iron accumulation, mitochondrial dysfunction, ATP reduction, synaptic loss, cognitive impairment. Each step in that chain is measurable and each was demonstrated experimentally in both directions, producing decline by increasing FTL1 and restoring function by reducing it.
The NADH Finding That Opens a Second Door
One of the more immediately interesting findings from the study involves a molecule called NADH. This is an electron carrier that fuels mitochondrial respiration, essentially a cofactor that helps mitochondria run the energy-producing process that FTL1 accumulation was disrupting. When the researchers supplemented FTL1-overexpressing mice with NADH, the damage was partially reversed. Neurons regrew their extensions. Memory performance improved.
Supplementing NADH, which fuels mitochondrial respiration, mitigated neurite loss and rescued memory in FTL1-overexpressing mice.
NADH is not a novel compound requiring decades of drug development. It is an endogenous molecule already present in every cell and available as a supplement. The finding does not mean NADH supplementation will reverse Alzheimer’s in humans. But it does mean the research identified a second entry point into the same pathway, one that bypasses FTL1 entirely and targets the downstream energy deficit directly. That is two potential therapeutic angles from a single study.
What This Looks Like in Human Brains
The study was conducted in mice, and that caveat matters. Every finding in mouse models requires human validation before it becomes clinically relevant, and the history of neuroscience is littered with interventions that worked brilliantly in rodents and failed in humans. Villeda’s team and independent commentators were careful to acknowledge this.
But the biological parallels are not trivial. FTL1 sits at the intersection of iron biology and neuronal metabolism. Iron dysregulation can damage mitochondria, undermine ATP production, and create oxidative stress. The work aligns with broader evidence that higher ferritin levels track with cognitive problems in older adults. Post-mortem analysis of human brains has found elevated hippocampal FTL1 in Alzheimer’s patients and people with mild cognitive impairment. The protein accumulates in the same region, in the same direction, in the same disease context. The mouse findings are not happening in isolation.
There is also a body of epidemiological research connecting elevated serum ferritin, a blood marker of iron storage, to cognitive decline in aging populations. High ferritin in older adults has been associated with poorer cognitive performance across multiple large studies. This research did not previously have a clear cellular mechanism to explain the connection. FTL1 provides one.
Why “Reversal” Is the Word That Changes Everything
There is an important distinction between the FTL1 finding and most of what gets published in the neuroscience of aging. The vast majority of research in this space focuses on prevention or delay. Eat well, exercise, sleep properly, keep your brain active, and you might slow the rate at which your cognitive function declines. That is useful advice, but it operates on the assumption that decline, once it has happened, cannot be undone.
The UCSF data does not fit that frame. When FTL1 was reduced in mice that were already old and already showing cognitive deficits, the deficits improved. The synapses that had weakened grew back. The memory performance that had declined recovered. The researchers did not find a way to slow the aging process in the hippocampus. They found a way to partially reverse it.
Villeda described the finding as suggesting brain aging is not necessarily permanent and may be reversible. That is a statement that would have been dismissed as unrealistic in most neuroscience contexts five years ago. The fact that it is coming from a Nature Aging paper with bidirectional experimental evidence behind it changes its weight considerably.
Human trials targeting FTL1 directly are realistically a decade or more away. The path from a mouse finding to an approved human therapy involves safety studies, dose optimization, delivery mechanism development, and clinical trials that take years each. But the identification of a specific, testable molecular target that can be reduced to produce measurable cognitive recovery is exactly the kind of finding that starts that process.
“It’s a hopeful time to be working on the biology of aging,” Villeda said.
Source:
Remesal, L., Lu, Y., Maynard, J.C., Burlingame, A.L., Villeda, S.A., et al. Targeting iron-associated protein Ftl1 in the brain of old mice improves age-related cognitive impairment. Nature Aging, 2025; 5: 1957–1969. DOI: 10.1038/s43587-025-00940-z https://www.nature.com/articles/s43587-025-00940-z